Lecture 5 Video 6
🧲 Protein Mobility & Dynamics from NMR Relaxation
(Based on Lecture 5 Video 6 )
This lecture explains how nuclear magnetic relaxation can be used to study protein motion and flexibility — from whole-molecule tumbling to tiny local fluctuations. The theory is heavy, but the concepts are powerful and very intuitive once broken down.
1️⃣ What Is Relaxation in NMR?
In NMR, we:
- Excite spins → create a non-equilibrium state
- Let nature restore equilibrium
- Measure how fast this happens
That return to equilibrium is called relaxation.
It is:
- A stochastic (random) process
- Occurring at a defined rate
- Responsible for signal decay
There are two main types:
🟢 T1 (Longitudinal Relaxation)
- Restores equilibrium magnetization
- Governs recovery along the magnetic field axis
🔵 T2 (Transverse Relaxation)
- Does not restore equilibrium
- Governs decay of observable signal in the transverse plane
⚠️ Important: Both T1 and T2 lead to disappearance of measurable magnetization.
2️⃣ Why Relaxation Tells Us About Protein Motion
Relaxation depends strongly on atomic mobility.
Proteins are not static objects — they move on many timescales:
| Motion Type | Timescale | Example |
|---|---|---|
| Overall tumbling | 1–10 ns | Whole protein rotating |
| Fast internal motion | ps | Methyl rotations |
| Intermediate motion | ns–µs | Loop flexibility |
| Slow motion | ns–ms | Helix breathing |
🌀 Correlation Time (τc)
Defined as:
Time required to rotate ~1 radian (57°) on average.
- Long τc → slow motion
- Short τc → fast motion
3️⃣ What Motions Can We Actually Measure?
✔ Overall tumbling → YES ✔ Fast internal mobility → YES ✖ Extremely fast (ps) → Hard to detect ✖ Very slow (ms–s) → Rarely measurable (real-time NMR possible but rare)
The most practical measurements:
- T1
- T2
- Heteronuclear NOE (¹H–¹⁵N)
4️⃣ The Model-Free (Lipari–Szabo) Approach
Instead of describing every motion separately, we compress mobility into:
- τM → overall molecular tumbling
- τe → internal motion correlation time
- Order parameter (S²) → rigidity measure
📊 The Order Parameter (S²)
Range: 0 → 1
| S² Value | Meaning |
|---|---|
| 1 | Completely rigid |
| ~0.9 | Very structured (secondary structure) |
| ~0.5 | Flexible loop |
| 0 | Completely decoupled from overall motion (rare) |
Interpretation:
- High S² → residue follows whole-protein tumbling
- Low S² → residue has independent internal motion
Typical values:
- Secondary structure → ~0.8–0.9
- Loops → ~0.4–0.6
Most residues fall between 0.5–0.9.
5️⃣ How T1 and T2 Depend on Molecular Size
T1 and T2 depend strongly on tumbling rate.
For small molecules (1–2 kDa):
- T1 shows a minimum
- T2 relatively long
For proteins (larger molecules):
- T1 increases
- T2 decreases
So for proteins:
The ratio T1/T2 increases as the molecule becomes larger or tumbles slower.
⚠️ Only valid for rigid residues (little internal motion).
Since ~80% of residues in a folded protein are structured, this works well.
6️⃣ Example: Detecting Dimerization via Relaxation
A small copper-binding protein was studied:
APO Form (no copper)
- T1 ≈ 0.4 s
- T2 ≈ 0.1 s
- Stable across residues
After Copper Addition
- T1 increased
- T2 decreased
- T1/T2 ratio increased strongly
Interpretation: 👉 Protein formed a dimer
Why?
- Copper needs 3 ligands
- Each protein provides only 2
- So 2 proteins bind one copper
Key point:
- No chemical shift changes
- No NOEs between monomers
- Symmetric complex → looks identical in shifts
But relaxation detects increased size → slower tumbling → dimer formation.
Relaxation can reveal what chemical shifts cannot.
7️⃣ Local Mobility: Heteronuclear NOE (¹H–¹⁵N)
Used to probe residue-specific flexibility.
Usually plotted as:
1 + NOE
Why? Technical definition reasons.
🧬 Example: Calmodulin

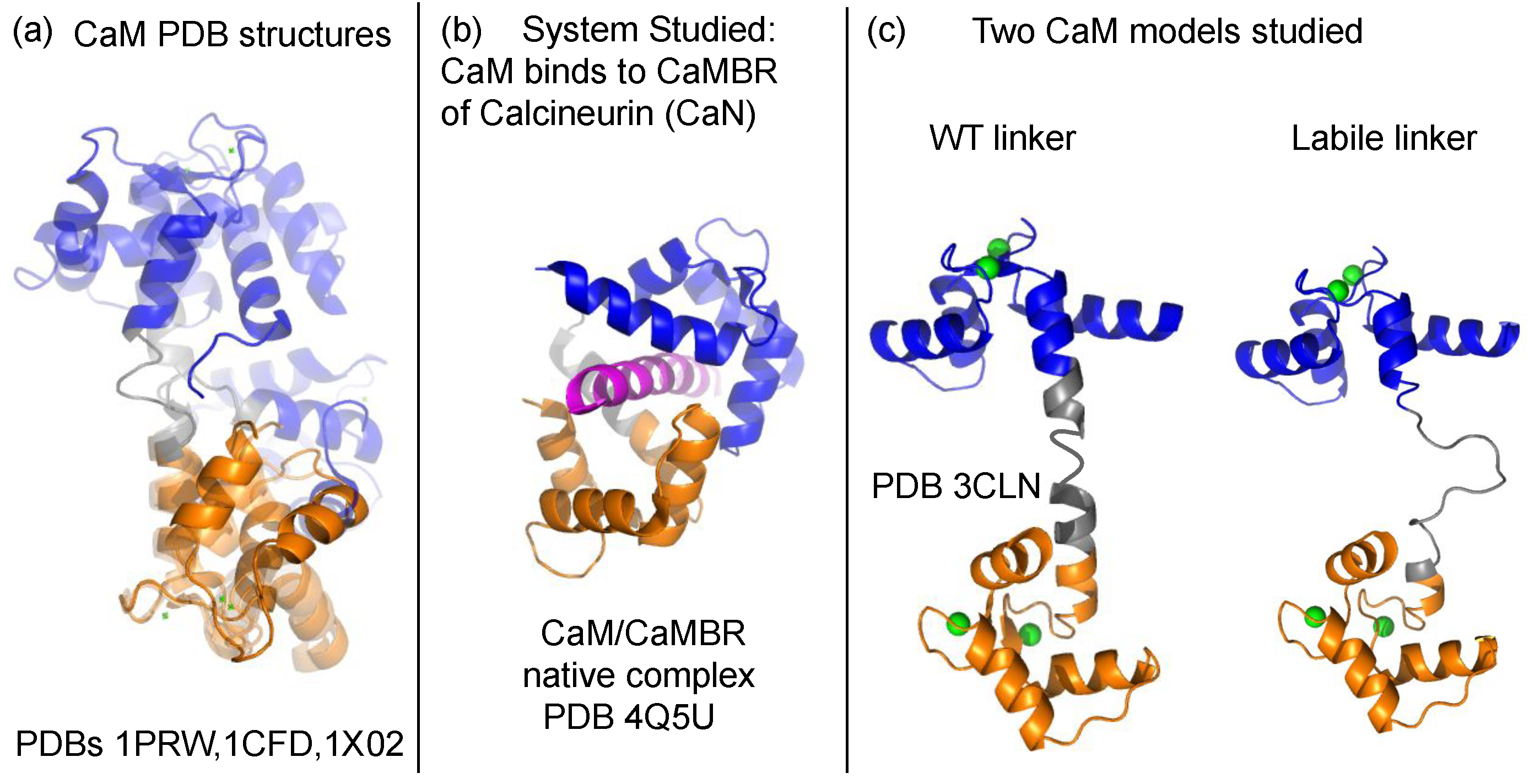


Calmodulin Structure:
- Well-folded N-lobe
- Well-folded C-lobe
- Flexible central linker
Superposition:
- N-lobe aligns well
- C-lobe aligns well
- Whole protein does not align → linker is flexible
NOE Results:
- High NOE → rigid lobes
- Low NOE → flexible linker
- N-terminus → highly flexible
- Loop regions → reduced NOE
So NOE maps flexibility along sequence.
8️⃣ Mutation Study: F141L in Calmodulin
Mutation:
- Phe141 → Leu
Result:
- NOE values reduced in opposite region
- Increased flexibility detected
- Very subtle mobility changes measurable
Interesting: Mutation at Phe89 also causes disease.
So: Relaxation can detect small mobility changes linked to pathology.
9️⃣ Calcium Removal Experiment
Without Ca²⁺:
- N-lobe remains folded
- C-lobe:
- Many residues invisible
- Remaining residues show reduced NOE
Interpretation: 👉 C-lobe fold destabilized 👉 Increased flexibility 👉 Structural disruption
Relaxation detects unfolding or partial destabilization.
🔟 Big Picture Takeaways
What Relaxation Can Tell You
✔ Overall tumbling rate ✔ Molecular size changes ✔ Dimerization ✔ Residue-specific flexibility ✔ Loop mobility ✔ Mutation-induced changes ✔ Folding stability
What It Cannot Easily Tell You
✖ Ultra-fast picosecond motions ✖ Very slow millisecond motions (hard, rare)
🧠 Conceptual Summary
Protein motion occurs at multiple timescales.
Relaxation translates motion into measurable parameters:
- T1 → energy recovery
- T2 → signal decay
- T1/T2 → molecular size & tumbling
- Heteronuclear NOE → local flexibility
- S² → rigidity index
Rigid residues:
- High S²
- High NOE
- Reflect global tumbling
Flexible residues:
- Lower S²
- Reduced NOE
- Decoupled from global motion
🎯 Why This Is Powerful
Relaxation allows you to detect:
- Dimer formation without chemical shift change
- Subtle mutation effects
- Loop flexibility
- Local unfolding
- Stability differences
It is one of the most sensitive tools for studying protein dynamics in solution.