Lesson 10 PPT
Protein structure
🧬 Molecular Simulation & Protein Structure Prediction
🔬 NMR vs X-ray Crystallography


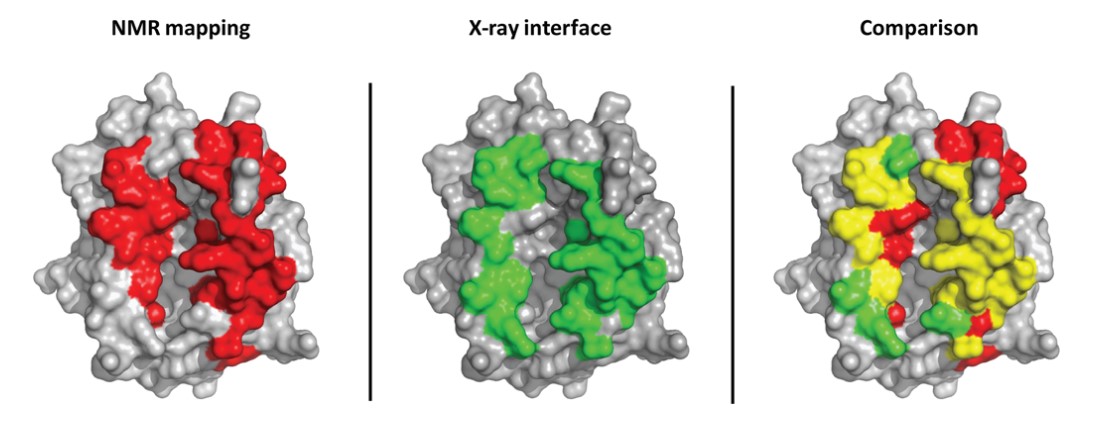

- NMR (Nuclear Magnetic Resonance)
- Produces an ensemble of structures (multiple conformations)
- Reflects protein flexibility in solution
- Useful for studying dynamic proteins
- X-ray crystallography
- Produces a single high-resolution structure
- Requires crystallization (non-physiological)
- Often misses flexible regions
👉 Key takeaway: NMR = dynamic + multiple conformations X-ray = static + high resolution
⚙️ Protein Folding Problem
- Proteins fold into structures that minimize free energy
- The folding problem = searching for global minimum energy
⚠️ Problem:
- Huge search space (ψ/φ backbone angles + side chains)
- Number of conformations grows exponentially
👉 This is why pure computational folding is hard
🧠 Protein Structure Prediction Methods
1. Ab initio (First principles)
- Uses physics only
- ❌ Too computationally expensive (not practical for large proteins)
2. Comparative Modeling
- Uses known structures as templates
3. Homology Modeling
- Based on sequence similarity
- If sequences are similar → structures are similar
4. Protein Threading
- Fits sequence into known structural folds, even with low similarity
🧵 Protein Threading
💡 Concept



- Place sequence onto known structure
- Evaluate how well it fits (energy scoring)
Why it works:
- Proteins adopt limited number of folds
- ~10 folds explain ~50% of structures
👉 Instead of searching infinite possibilities → reuse known folds
⚙️ Threading Components
- Structural template database
- Energy/scoring function
- Alignment algorithm
- Reliability assessment
📊 Scoring (Punctuation) Functions
Key factors:
- Solvation potentials (buried vs exposed residues)
- Contact potentials
- Secondary structure agreement
- Accessibility predictions
🔥 Contact Potential (Important!)
Uses Boltzmann principle:
- Favorable contacts occur more frequently
- Energy is derived from observed frequencies
👉 Translation:
- Common interactions → energetically favorable
🧬 Sequence Profiles + Secondary Structure
- Combines:
- Evolutionary info (profiles)
- Predicted secondary structure
👉 Improves accuracy significantly
📊 Post-processing & Evaluation
- Filter bad models
- Combine additional data
- Benchmark using CASP experiments
🤖 AlphaFold (Deep Learning Revolution)
🧠 Overview
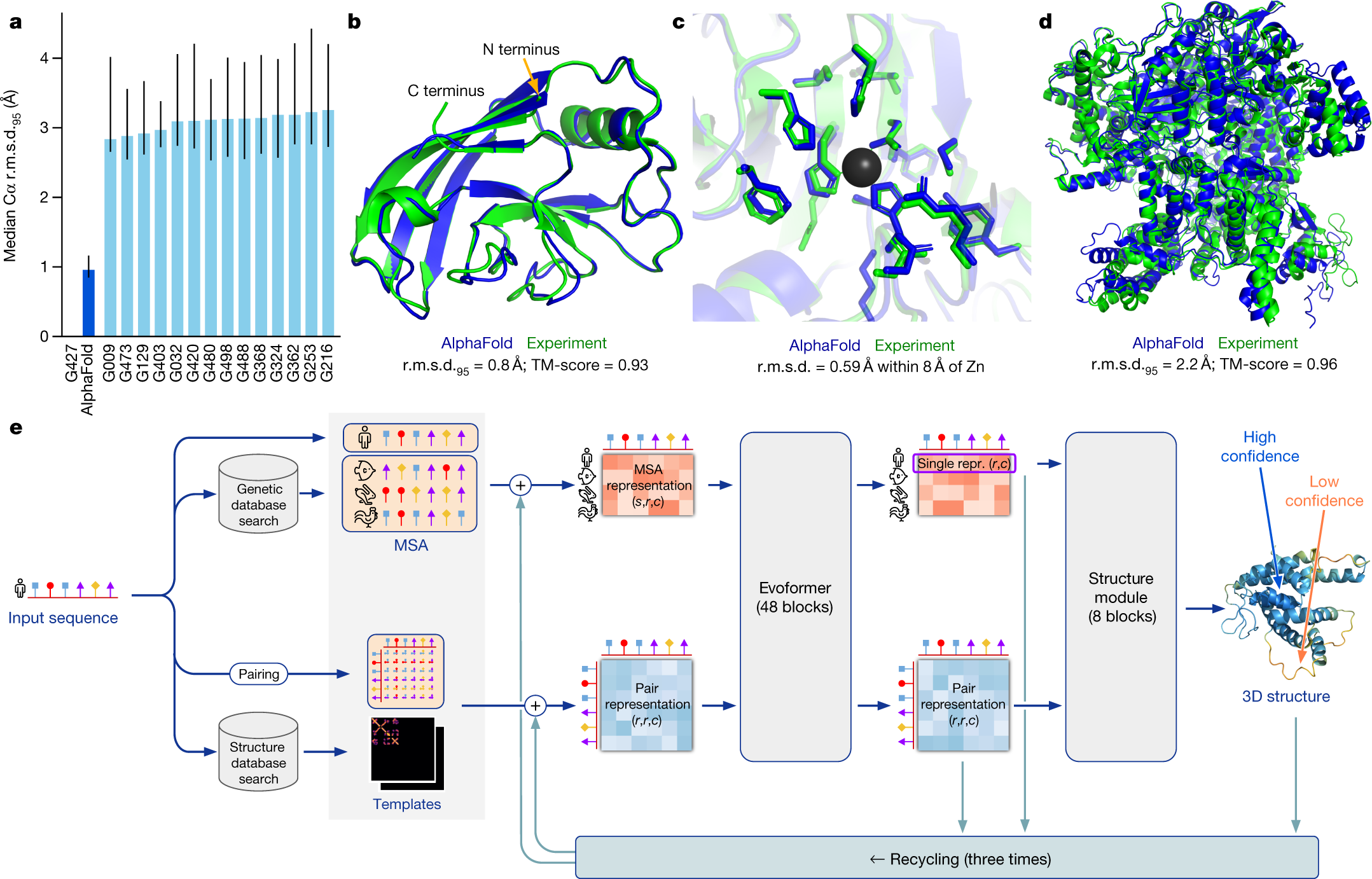

- Uses deep learning + evolutionary data
- Predicts 3D structure from sequence
🧬 Key Components
1. Input
- Amino acid sequence
2. MSA (Multiple Sequence Alignment)
- Detects:
- conserved residues
- co-evolution (residues interacting)
👉 If two residues mutate together → likely interact
3. Evoformer (Core engine)
- Transformer-based
- Learns:
- long-range interactions
- structural constraints
4. Structure Module
- Converts predictions into 3D coordinates
- Uses Invariant Point Attention (IPA)
👉 Important:
- Handles spatial geometry properly
5. Output
- Full atomic model
- Confidence metrics:
- pLDDT → per residue confidence
- PAE → domain relationship uncertainty
🧪 Molecular Docking
🔑 What is Docking?



- Predicts how a ligand binds to a protein
⚠️ Important:
- Does NOT directly predict bioactivity
🔄 Docking Theories
- Lock-and-key → rigid fit
- Induced fit → protein adapts
⚙️ Two Main Steps
1. Sampling
- Try many ligand conformations
2. Scoring
- Rank based on binding energy
🧬 Types of Docking
- Protein–protein
- Protein–ligand
- Protein–nucleotide
🎯 Applications
- Reproduce known binding modes
- Predict binding of known ligands
- Estimate binding affinities
- Virtual screening (drug discovery)
⚙️ Algorithms
- Use:
- conformational search methods
- scoring functions
🔍 Docking Workflow
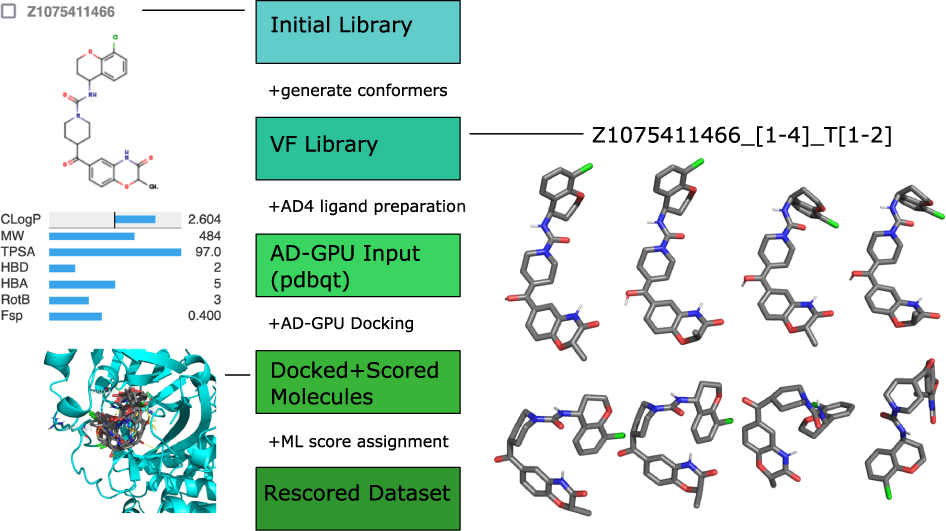

Typical steps:
- Prepare protein + ligand
- Define binding site
- Generate conformations
- Score poses
- Rank results
🧪 Validation Methods
🔁 Redocking
- Dock ligand back into known structure
- Tests accuracy
🔄 Cross-docking
- Dock ligand into different structures
- Tests robustness
📈 ROC Curve (Image slide explanation)




- Measures model performance
- AUC (Area Under Curve):
- 1.0 = perfect
- 0.5 = random
👉 Used in virtual screening
🔬 Binding Energy Example
- Example: -10.10 kcal/mol
- More negative = stronger binding
🧾 Docking Score Table
- Compares different ligand poses
- Used to select best candidate
⚠️ Types of Docking (Advanced)
- Covalent docking → ligand forms bond
- Blind docking → unknown binding site
- Reverse docking → one ligand vs many proteins
⚖️ Pros & Cons of Docking
✅ Pros
- Fast screening
- Cost-effective
- Useful for drug discovery
❌ Cons
- Scoring functions imperfect
- Protein flexibility limited
- Does not guarantee biological activity
🧠 Big Picture Summary
- Protein structure prediction evolved from:
- ❌ physics-based → too slow
- ✅ template-based → useful
- 🚀 AI-based (AlphaFold) → breakthrough
- Docking:
- Predicts binding mode, not activity
- Depends heavily on sampling + scoring quality
🔥 Key Concept Connections (Important for Exams)
- Threading vs Homology modeling
- Homology → sequence similarity
- Threading → structure similarity
- AlphaFold vs Docking
- AlphaFold → structure prediction
- Docking → interaction prediction
- Energy principle
- Folding → minimize energy
- Docking → optimize binding energy
Quiz
Score: 0/30 (0%)