Day 10 part 2
🧬 Molecular Docking — Full Conceptual Overview
🧠 What is Molecular Docking?
Molecular docking is a computational method used to predict how a molecule (ligand) binds to a protein (target).
- Goal:
- Find binding position (pose)
- Estimate binding strength (affinity)
- Predict stability of the complex
📌 Key idea:
“Which ligand fits best into the protein, and how strongly?”
⚠️ Important Limitation (Often Misunderstood)
Docking is NOT designed for large molecules (e.g., polymers).
Why?
- Only a small region (active site) is considered
- Large molecules extend outside → unrealistic scoring
✔️ Correct approach:
- Break polymers into small fragments before docking
🔑 Core Concept: Lock-and-Key vs Induced Fit
🔒 Rigid docking (Lock-and-key)
- Protein = fixed
- Ligand = fixed
- Works like LEGO blocks
➡️ If geometry doesn’t match → no binding
🔄 Flexible docking (Induced fit)
- Ligand (and sometimes protein) can change shape
- More realistic but computationally heavier
🔄 Sampling Conformations (Your Question 1)
✔️ Your understanding is correct.
What happens:
- Place ligand in active site
- Rotate flexible bonds step-by-step
- Generate many conformations
- Evaluate each one
➡️ “Which conformation interacts best?”
📌 Important nuance:
- Rotation ignores physical barriers (not realistic movement)
- It just tests possibilities
🧮 Scoring Function → Ranking
Each conformation gets a score based on:
- van der Waals interactions
- electrostatics
- solvation energy
➡️ Final goal:
Lowest energy = best binding
✔️ Yes, you are right: 👉 We aim for minimum energy
🧬 Example: HIV Protease (Your Question 2)
HIV protease

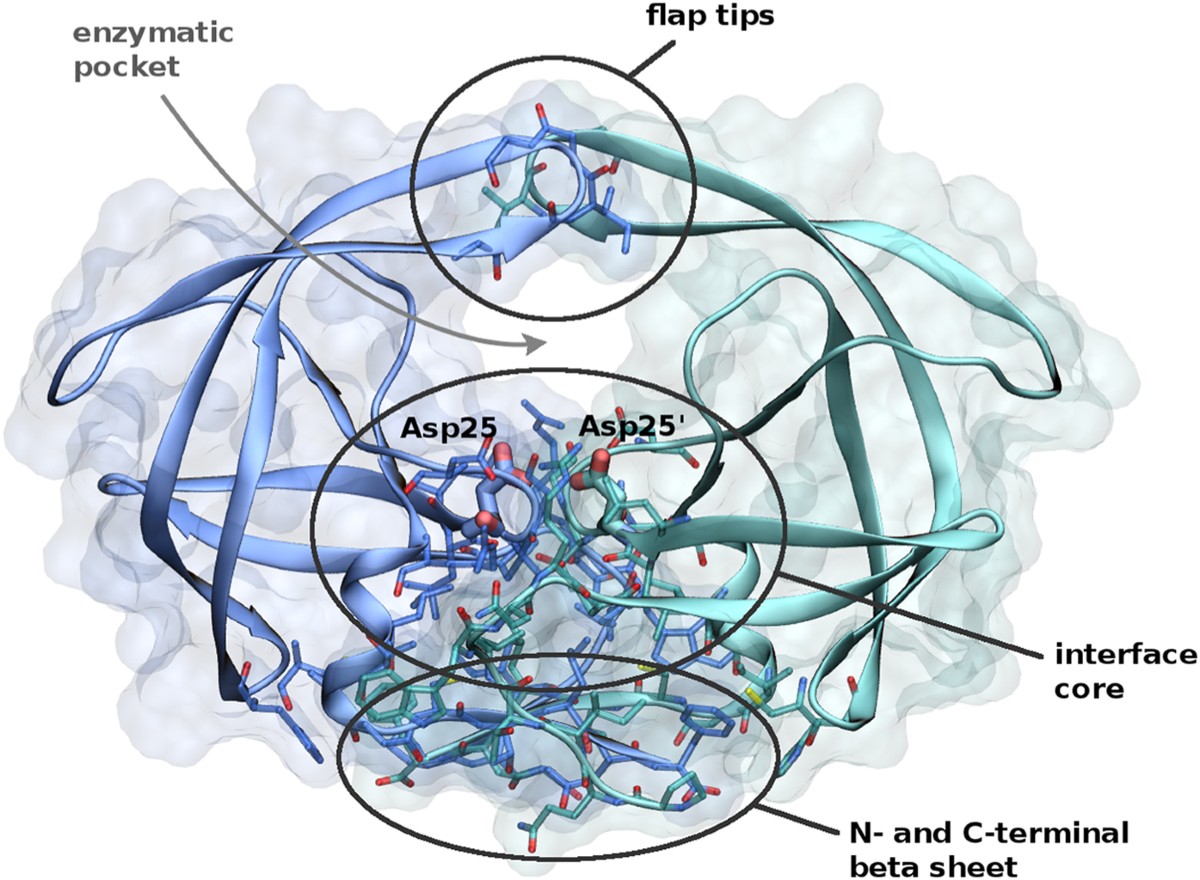
Why it’s important:
- Target for anti-HIV drugs
- Inhibitors block enzyme → virus cannot mature
Key problem:
HIV mutates very fast
➡️ Implication:
- Binding site changes → drugs stop working
- Docking must account for mutations
✔️ Insight: Docking is used to design new inhibitors faster than mutations evolve
🧬 Protein–Protein Docking (Your Question 3)
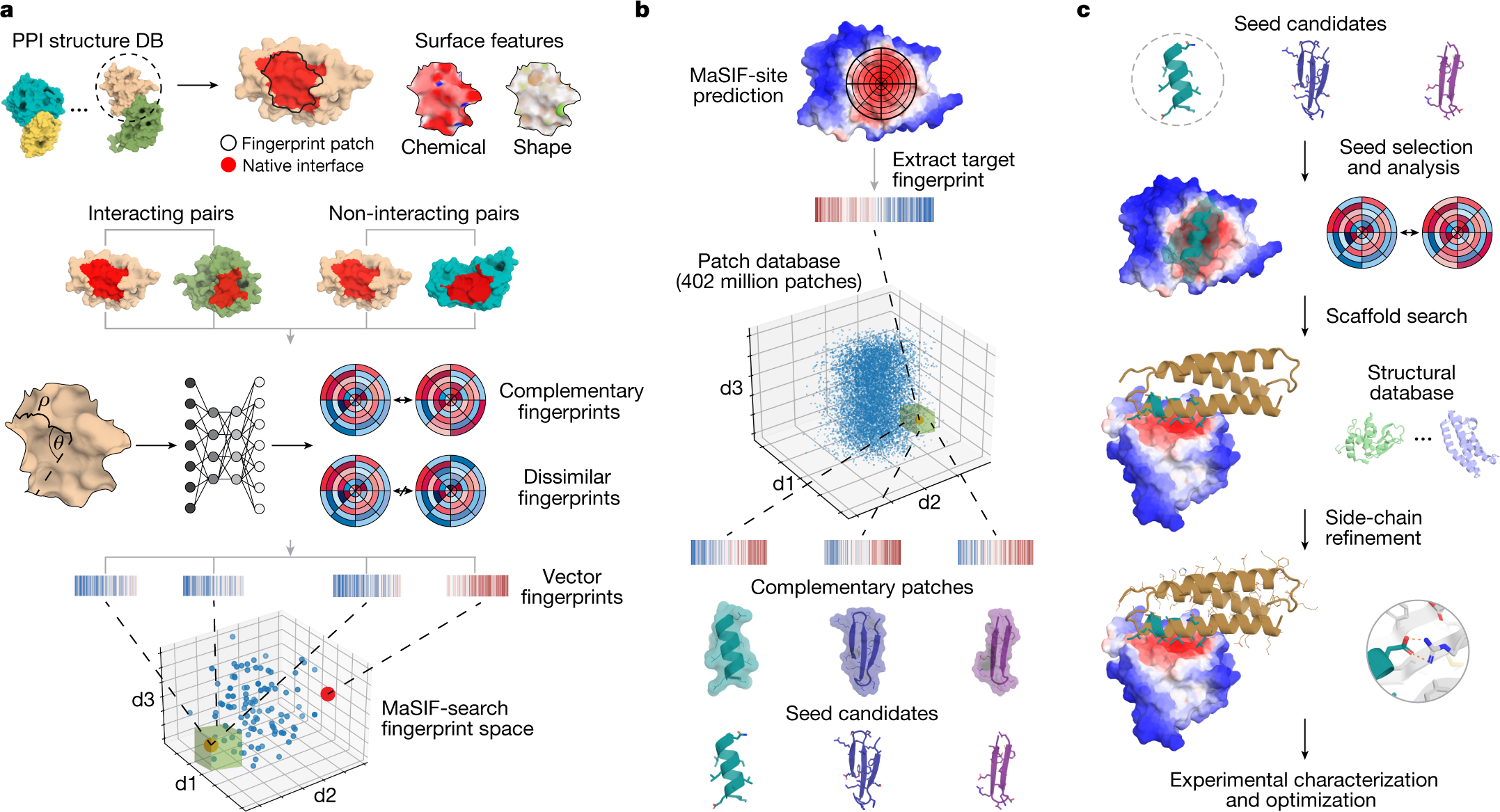


What it means:
- Predict how two proteins interact
Applications:
- Protein complexes
- Signaling pathways
- Enzyme regulation
Difference vs small molecule docking:
- Larger interfaces
- More complex geometry
- More flexibility
🧪 Reproducing Binding Mode (Your Question 4)
What this means:
You already have:
- Experimental structure (X-ray / NMR)
Goal:
- Check if docking can reproduce the same binding pose
📌 Why important:
- Validates docking method
- Confirms scoring function reliability
✔️ If docking ≈ crystal structure → method is trustworthy
🧩 Fragment-Based Docking (Your Question 5)
✔️ Your understanding is correct.
Process:
- Break ligand into fragments
- Dock fragments individually
- Recombine best fragments
➡️ Build optimized molecule step-by-step
📌 Advantage:
- Explores chemical space efficiently
⚙️ Docking Workflow (Corrected & Expanded)
1️⃣ Protein & Ligand Selection
- Need:
- Protein structure (PDB)
- Ligand structure (mol2, etc.)
2️⃣ Protein Preparation (Your Question)
Why protonate?
X-ray structures do not include hydrogens
➡️ But hydrogen atoms are essential for:
- Hydrogen bonding
- Electrostatics
So you must: ✔️ Add hydrogens ✔️ Assign correct protonation states
⚠️ Critical Issue: Histidine (Your Question)
Histidine
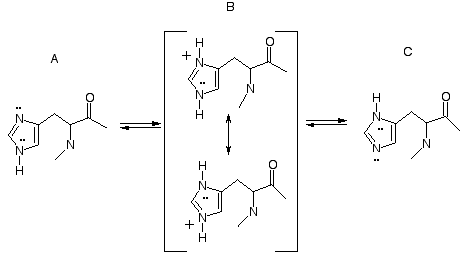

Histidine can be:
- Protonated at Nδ1
- Protonated at Nε2
- Both (charged)
- None
Why this matters:
- Wrong protonation → wrong hydrogen bonds
- Can completely ruin docking results
✔️ Key rule:
Protonation must match local environment of active site
Additional Preparation Steps
- Remove crystal waters
- Define active site residues
📦 Binding Box (Docking Box)
What is it?
A 3D region where docking occurs
Trade-off:
- Too small → miss interactions
- Too large → inefficient & noisy
✔️ Best:
Small but includes all active residues
🎯 Docking Types
🔍 Blind Docking (Your Question)
✔️ Your description is correct.
- Dock ligand across entire protein surface
- Many simulations → map binding hotspots
➡️ Output:
- Clusters of preferred binding sites
🎯 Focused Docking
- Restrict docking to known active site
✔️ More accurate, faster
🔄 Reverse Docking (Your Question)
✔️ Correct idea.
Instead of:
- Many ligands → 1 protein
You do:
- 1 ligand → many proteins
➡️ Find:
Which protein binds best?
🔁 Redocking (Your Question)
What it means:
Take a ligand from:
- Known crystal structure
Then:
- Remove it
- Dock it again
➡️ Compare predicted vs experimental pose
✔️ Purpose:
- Evaluate docking accuracy ("docking power")
🔬 Docking + MD Simulation
Docking gives:
- Fast prediction
But:
- Not always realistic
So: ➡️ Run Molecular Dynamics (MD)
Check:
- Does ligand stay bound?
- Or diffuse away?
✔️ Key insight:
Good docking ≠ stable complex
⚖️ Energy Principle (Core Concept)
✔️ You are correct:
Systems aim for minimum energy
Docking searches:
- Many conformations
- Finds lowest-energy state
⚠️ Pros & Cons
✅ Advantages
- Fast
- Good for screening large libraries
- Low computational cost
❌ Limitations
- Accuracy depends on:
- Protonation
- Flexibility assumptions
- Can give false positives
- Needs post-validation (MD)
🔄 Docking vs MD (Important Distinction)
Docking:
- Samples conformations artificially
- Ignores energy barriers
MD:
- Follows real physics
- Includes time evolution
✔️ Key difference:
Docking jumps between states, MD simulates transitions
🧠 Final Takeaways
- Docking = pose prediction + scoring
- Best pose = lowest energy
- Preparation (especially protonation!) is critical
- Flexible docking is more realistic but harder
- Always validate with MD or experiment