PPT 7
Protein Purification & Chromatography — Full Educational Summary
Page 1 — Big Picture: What is protein purification? 🧪
This opening slide gives you the full workflow.
The figure on the left shows:
Preparation → Capture → Intermediate purification → Polishing
This is the standard purification pipeline.
Purity increases step by step.
Think of it like filtering sand from water:
- first remove rocks
- then remove smaller particles
- finally polish until crystal clear
For proteins, the same logic applies.
You begin with a complex biological mixture:
- cell lysate
- serum
- tissue extract
- bacterial culture
containing:
- your target protein
- thousands of other proteins
- DNA/RNA
- lipids
- salts
- debris
The SDS-PAGE image at the bottom shows this beautifully:
- many bands = crude mixture
- fewer bands = purification progressing
- one band = high purity protein
This is one of the most important visual concepts in purification.
Page 2 — Purification strategy 🧠
This page is extremely important.
Before purifying anything, you must ask:
What is the protein used for?
This determines required purity.
Examples:
Therapeutic use
Needs extremely high purity because contaminants can be dangerous
Often >99%
Structural biology
For:
- X-ray crystallography
- cryo-EM
- NMR
Needs very high purity and homogeneity
Usually 95–99%
Activity assay
Sometimes moderate purity is enough if contaminants do not interfere
Important strategy rules
1. Keep steps minimal
Each step loses protein.
Even 90% yield per step becomes:
- 1 step → 90%
- 2 steps → 81%
- 5 steps → 59%
- 8 steps → 43%
The graph on the slide shows this exponential loss.
This is a key concept.
Many purification failures happen because too many steps are used.
2. Use orthogonal methods
This means each step should separate by different properties.
For example:
- charge → ion exchange
- size → size exclusion
- binding specificity → affinity
This greatly improves purity.
3. Avoid dilution
Dilution can:
- destabilize proteins
- reduce concentration
- increase losses
Very important in lab work.
Page 3 — The CIPP strategy ⭐
This is one of the most important concepts.
C = Capture
Goal:
- isolate
- concentrate
- stabilize
This is your first major purification step.
Example:
His-tag affinity column
This quickly pulls your protein out of a complex lysate.
I = Intermediate purification
Remove most contaminants.
This is often:
- ion exchange
- HIC
P = Polishing
Final cleanup.
Removes:
- aggregates
- closely related contaminants
- oligomeric species
Often done with:
- size exclusion chromatography (SEC)
This gives highly pure protein.
Page 4 — What must you know before purification? 🔬
This slide is very practical.
Required purity
Depends on experiment.
Very important.
Examples from slide:
- therapeutic → >99%
- crystallography → 95–99%
- antibody antigen → <95% can be enough
Analytical methods
You must monitor purification continuously.
SDS-PAGE
Fastest and most common
Shows:
- molecular weight
- approximate purity
MS
Mass spectrometry confirms exact identity
Activity assay
Sometimes purity alone is not enough.
Protein must still be functional.
This is especially important for enzymes.
Stability properties
This is critical.
You must know whether protein tolerates:
- temperature
- pH
- salt
- detergent
- redox conditions
This directly affects buffer design.
Page 5 — Sample extraction and clarification 🧫
Before chromatography, the sample must be clean.
The goal:
remove anything that can clog the column
Examples:
- cell debris
- membranes
- precipitates
- DNA clumps
Methods
Centrifugation
Spins debris down.
Supernatant contains soluble proteins.
Filtration
Removes particles.
Often:
0.22 µm filter
Very common before FPLC.
Precipitation
This is important.
Protein solubility can be manipulated.
Common agents:
- ammonium sulfate
- PEG
The SDS-PAGE image shows selective enrichment.
Bands become fewer after precipitation.
This means some proteins precipitated while others remained soluble.
Very important pre-purification step.
Page 6 — Ammonium sulfate precipitation 🧂
Very exam-relevant.
This table tells how many grams of salt to add.
Example from slide:
To reach 40% saturation
Add 243 g/L
Then from 40% to 70%
Add 205 g/L
This is called fractional precipitation.
Extremely useful.
Different proteins precipitate at different salt concentrations.
This is based on salting out.
Salt removes water from protein surfaces, reducing solubility.
Page 7–13 — Introduction to chromatography 🚰
This section explains the general principle.
Core principle
Chromatography separates molecules by repeated partitioning between:
- stationary phase = matrix
- mobile phase = buffer


Proteins interact differently with the matrix.
Some move faster.
Some slower.
This creates separation.
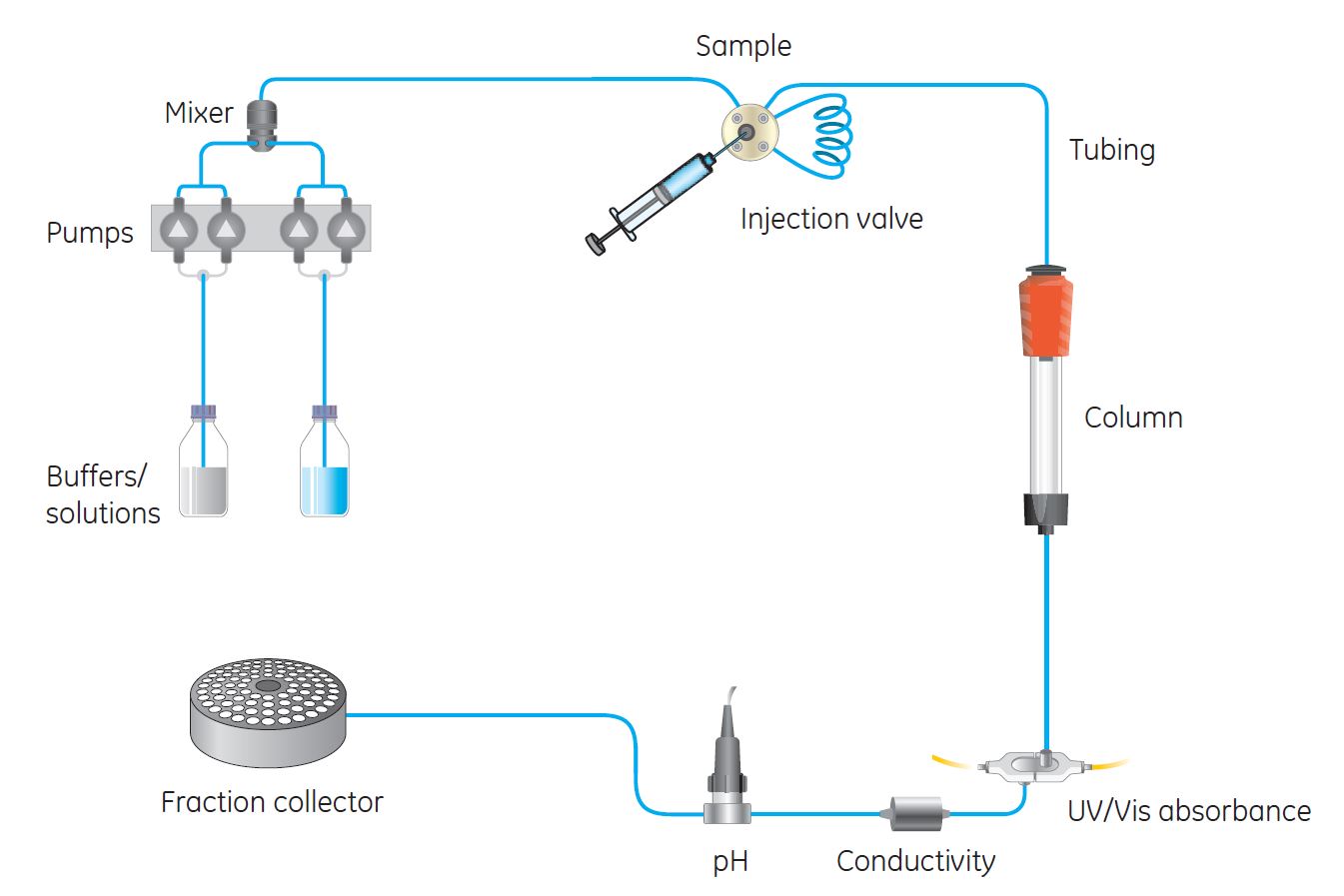
Instrument setup
The system images show:
- solvent reservoirs
- pumps
- injector
- column
- detector
- fraction collector
This is exactly what you see in an FPLC/HPLC system.
The detector commonly measures:
A280
Protein absorbance due to:
- tryptophan
- tyrosine
Chromatographic peaks
Very important concept.
Each peak corresponds to a separated protein species.
The peak position tells:
when it eluted
The peak area tells:
how much protein
Usually Gaussian-shaped.
This is important for fraction collection.
Affinity Chromatography (Pages 14–32) 🎯
This is probably the most important purification method.
Principle
Uses specific biological recognition.
Example:
protein binds ligand specifically
Examples:
- enzyme ↔ cofactor
- antibody ↔ antigen
- His-tag ↔ Ni²⁺
- GST ↔ glutathione
How it works
- ligand immobilized on beads
- target binds
- impurities wash away
- target specifically eluted
This gives extremely high purity in one step.
Binding equation
Very important:
heta = rac{L}{K_D + L}
Where:
- θ = saturation fraction
- KD = dissociation constant
- L = ligand concentration
heta = rac{L}{K_D + L}
Lower KD = stronger binding
This is exactly the same concept you’ve worked with before in binding curves.
Leakage concept
Pages 21–22 explain something very important.
If KD is too high:
protein leaks off during washing
For efficient purification:
binding KD must be low during loading
but high during elution
This often requires 1000-fold change
Very important concept.
Elution methods
Nonspecific elution
Change:
- pH
- salt
- chaotrope
This weakens binding
Specific elution
Add competing ligand
Example:
glucose competes off glucose-binding protein
This is usually gentler.
His-tag purification ⭐
This is extremely important.
Nickel / Ni-NTA purification.
His residues coordinate nickel.
This is immobilized metal affinity chromatography (IMAC).
Common for recombinant proteins.
Elution
Usually with:
imidazole
because it competes with histidine
This is likely one of the most useful methods you’ll use in protein biochemistry.
Ion Exchange Chromatography (Pages 33–52) ⚡
Very important.
Separates proteins by net charge.
Basic principle
Opposite charges attract.
Protein charge depends on pH relative to pI.
Above pI
protein is negative
binds anion exchanger
Below pI
protein is positive
binds cation exchanger
This is very important.
Elution
Usually by increasing salt.
Example:
NaCl gradient
Salt ions compete with protein for binding sites.
This causes elution.
The stronger the protein binds, the higher salt needed.
Gradient elution
The slides show:
- linear gradient
- complex gradient
- step gradient
This is extremely common in FPLC.
A salt gradient allows fine separation.
pH effect
pH changes protein charge.
Therefore pH strongly affects retention.
This is why buffer choice is critical.
Hydrophobic Interaction Chromatography (Pages 61–67) 💧
This is often confusing but very important.
Proteins contain hydrophobic surface patches.
These bind hydrophobic matrices.
High salt promotes binding
This is opposite of ion exchange.
High salt strengthens hydrophobic interaction.
Why?
Because salt strips water away from hydrophobic surfaces.
This exposes hydrophobic patches.
Then they bind matrix.
Elution
Decrease salt concentration.
Less hydrophobic interaction → protein elutes.
Hofmeister series
Very important concept.
Some salts are better at salting out proteins.
Especially:
(NH_4)_2SO_4
This is why ammonium sulfate is used so much.
Size Exclusion Chromatography (Pages 68–84) 📏
This is one of the easiest methods conceptually.
Separates by size.
Principle
Porous beads contain holes.
Small proteins enter pores.
Large proteins cannot.
Result
Large proteins
elute first
Small proteins
elute later
This is extremely important.
Students often initially think the opposite.
Hydrodynamic size
Important detail:
separation depends on shape + size, not molecular weight alone
This is why unfolded proteins appear larger.
The later slides explicitly show this.
Very important for interpreting SEC.
Denatured proteins
Pages 81–84 show denatured proteins appearing larger.
This is because unfolded proteins behave like expanded coils.
This is a key biophysical concept.
Very relevant to your background.
Final pages — Strategy + exercises ✨
The lecture ends by bringing everything together.
The key idea is:
choose purification method based on protein property
| Property | Method |
|---|---|
| Specific binding | Affinity |
| Charge | Ion exchange |
| Hydrophobicity | HIC |
| Size | SEC |
This is the central takeaway.
High-yield exam summary 🎯
Remember this table:
| Method | Separates by |
|---|---|
| Affinity | specific binding |
| IEX | charge |
| HIC | hydrophobicity |
| SEC | size |