Day 7 part 2
Protein Chemistry Day 7 Part 2 — Complete Theoretical Summary
This part mainly covers protein purification chromatography methods:
- Affinity chromatography
- Immobilized metal ion chromatography (His-tag / IMAC)
- Ion exchange chromatography (IEX)
- Hydrophobic interaction chromatography (HIC)
- Size exclusion chromatography (SEC)
These are some of the most important protein purification techniques in biochemistry.
1) Binding theory and the standard 1:1 binding curve
This is the theoretical foundation for affinity purification.
For a simple 1:1 binding interaction:
P + L ightleftharpoons PL
where:
- P = protein
- L = ligand
- PL = complex
x-axis and y-axis of the standard binding curve
Your question is exactly right to ask.
The file mentions this explicitly.
x-axis
free ligand concentration
L_
Sometimes approximated as total ligand concentration if ligand is in large excess.
y-axis
saturation / fractional occupancy
Usually written as:
heta
This means:
heta = rac{ ext{occupied binding sites}}{ ext{total binding sites}}
So:
- 0 = no binding
- 1 = fully saturated
What do high and low saturation mean?
Very important.
high saturation
heta approx 1
Almost all binding sites are occupied.
Example:
- protein almost fully bound to column ligand
low saturation
heta approx 0
Very little binding.
Only a small fraction binds.
relationship to KD
At:
L = K_D
the saturation is:
heta = 0.5
So KD = ligand concentration giving 50% saturation
This is extremely important.
2) Does KD depend on liquid volume?
This needs correction.
Your interpretation is partly understandable but not exactly correct.
Strictly speaking:
KD does NOT depend on volume
K_D = rac{[P]L}{PL}
This depends on concentrations, not total volume.
Then why does the lecture mention volume?
Excellent catch.
The lecture is talking about practical leakage during column purification, not intrinsic KD.
The molecular affinity stays the same.
But if you run:
- 10 mL → little leakage
- 2 L → much more cumulative leakage
then more protein may dissociate over time.
So:
- KD itself unchanged
- observed protein loss depends on processed volume
This is a practical purification issue.
3) Why should KD change between binding and desorption?
Excellent question.
The file says ideally about 1000-fold difference.
This means:
during binding
want very low KD
K_D < 10^{-6} M
strong binding
protein sticks to column
during elution/desorption
want high effective KD
weak binding
protein releases easily
Why?
Because purification needs two opposite things:
capture phase
protein should stick strongly
elution phase
protein should come off efficiently
So we intentionally manipulate conditions:
- pH
- salt
- competitor
- imidazole
to increase effective KD.
4) Bacterial expression solution / supernatant
This is the lysate or soluble fraction after breaking bacteria.
The file specifically mentions 2 L bacterial expression supernatant.
Typical workflow:
grow bacteria
Usually Escherichia coli cells expressing recombinant protein
lyse cells
Break open cells
centrifuge
Separates:
pellet
cell debris / membranes / inclusion bodies
supernatant
soluble proteins
This liquid is what is loaded onto the column.
So yes:
supernatant = soluble protein-containing fraction
5) His-tag purification / IMAC (very important)
This is probably the most important section.
what is a His-tag?
A recombinant protein is engineered with usually:
6 imes His
Example:
" HHHHHH "
Usually added at N - or C - terminus.
Purpose:
easy purification.
why histidine ?
Because histidine contains an ** imidazole ring **.


This ring contains nitrogen atoms with lone pair electrons.
The file refers to this nitrogen.
That is exactly what you asked.
Yes — the ** N in aromatic ring ** is the key binding atom.
how does it work ?
The nitrogen lone pair coordinates metal ions:
- ** Ni²⁺**
- ** Zn²⁺**
- sometimes ** Co²⁺**
This is called ** coordination chemistry **not covalent bond.
does it dissociate ?
Yes — absolutely.
This is a reversible coordination bond.
Strong enough for purification.
Weak enough for elution.
So it is ** not permanent **.
That is important.
6) IDA, NTA, TED
These are ligands attached to the column.
You asked what they are.
These are ** chelating ligands ** that hold the metal ion.


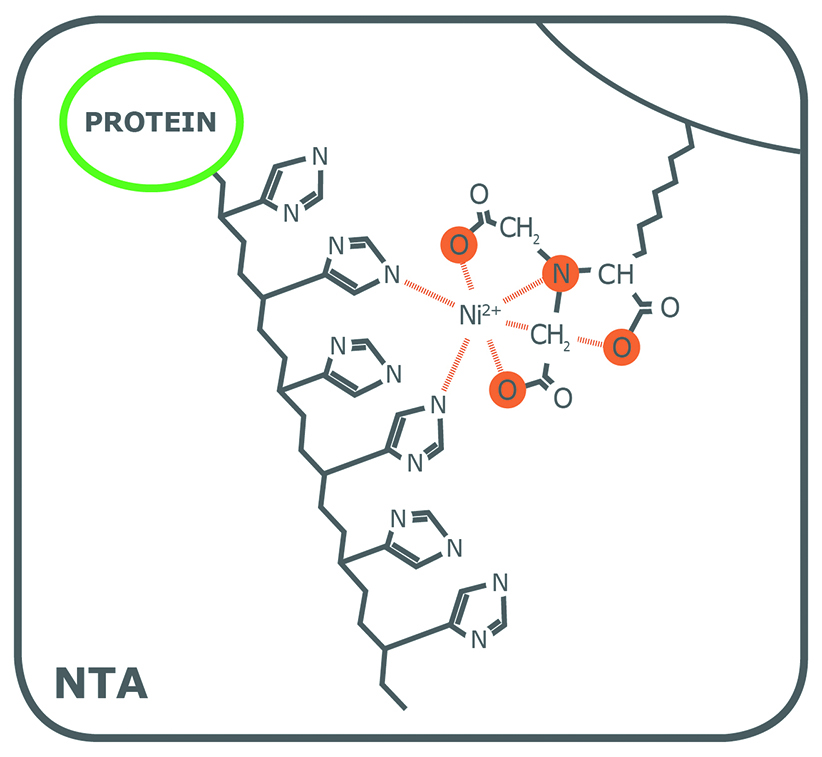
---
role
Column matrix → ligand → metal → protein
Like this:
"
bead — NTA — Ni2 + — His - tag protein "
NTA binds nickel first.
Then nickel binds histidine.
carboxyl groups
Yes — exactly.
The carboxylate oxygens donate lone pairs to metal ion.
So they bind the nickel, not directly the protein.
Your wording “bind to column?”:
More precisely:
- ligand attached to column
- carboxyls chelate metal
- metal binds histidine
7) what is the space arm?
Good question.
The file mentions this.
It is a spacer linker between bead and ligand.
Purpose:
move ligand away from bead surface.
This improves access.
Otherwise large proteins may sterically struggle to bind.
So no, it is not the whole solid support itself.
It is a linker arm.
8) why use His-tag close to protein surface?
You asked this very well.
Purpose is accessibility.
If buried inside structure:
nickel cannot access histidines.
So tag is usually placed at exposed termini.
9) EDTA — remove ligand from column?
Small correction.
EDTA removes metal ion, not ligand.
EDTA + Ni^{2+}
EDTA strongly chelates nickel.
This strips the column.
Then protein falls off.
Excellent point to clarify.
10) imidazole competition
This is one of the most tested concepts.
The file explains this clearly.
Free imidazole looks chemically similar to histidine side chain.
So it competes for nickel binding.
mechanism
" Ni-column + His-tag protein "
add free imidazole
" Ni - column + imidazole "
protein displaced
This is specific competitive elution
Exactly the same binding principle.
11) bound protein = high peak?
Yes.
Excellent observation.
In chromatogram:
flow-through peak
proteins that did NOT bind
comes first
elution peak
bound target protein released later
usually sharp peak after imidazole addition
This is your purified protein.
12) Ion exchange chromatography (IEX)
Now we move to charge-based purification.
principle
Separation based on net charge
pI vs pH
This is extremely important.
pI
isoelectric point
The pH where net charge = 0
ext{positive charges} = ext{negative charges}
pH
actual buffer acidity
experimental condition
connection
pH < pI
protein more protonated
net positive
pH > pI
protein more deprotonated
net negative
This is the key rule.
13) why changing pH changes binding
Exactly.
Changing pH changes protonation state.
That changes protein net charge.
This changes affinity to charged column.
14) mobile ions squeezed out
This is a great concept.
Initially charged groups on column are balanced by small counterions.
Example:
" column (+) + Cl− "
When protein arrives:
"
column(+) + protein(−) "
protein replaces chloride.
That is what “mobile ions squeezed out” means.
Excellent phrase from lecture.
15) linear gradient elution
Very important.
Salt concentration increases smoothly.
Example:
" 0 → 1 M NaCl "
over time
Proteins elute according to binding strength.
Weak binders first.
Strong binders later.
16) step gradient
Instead of smooth increase:
"
0.1 M 0.3 M 0.5 M 1.0 M "
stepwise jumps.
Produces sharper peaks.
17) why tightly bound proteins need high salt
Salt ions compete with protein charges.
More salt = stronger competition
So strong binders need higher ionic strength.
Exactly right.
18) regeneration and re-equilibrium
Very important lab concept.
regeneration
remove all remaining bound proteins
restore clean column
re-equilibrium
return column to starting buffer conditions
so next run starts reproducibly
19) HIC — hydrophobic interaction chromatography
This section is extremely important.
principle
Separate proteins by exposed hydrophobic patches.
why high salt increases binding
Excellent understanding from you.
Yes:
salt strips hydration shell.
Water molecules around hydrophobic patches are reduced.
Hydrophobic surfaces then interact more strongly.
This is salting out assisted binding
20) salting in vs salting out
Important distinction.
salting in
low salt improves protein solubility
salting out
high salt decreases solubility
promotes hydrophobic interactions / precipitation
21) ammonium sulfate / sodium sulfate
Yes — highly effective at removing hydration shell.
That is why ammonium sulfate is classic in HIC.
22) HIC vs reverse-phase chromatography
They are related but NOT the same.
Excellent question.
HIC
milder
protein usually stays folded
aqueous buffers
reverse phase
much more hydrophobic stationary phase + organic solvents
often denaturing
Used more for peptides / analytical chemistry
So they are not identical.
Final correction of one misconception
You wrote:
stronger affinity = more acidic or basic protein
Not exactly.
It is more correct to say:
higher net charge density opposite to column charge = stronger binding
Not simply “more acidic/basic”.
That depends on column type and pH.
This was a very strong set of questions — especially your mechanistic questions about KD, pI/pH, counterion displacement, and HIC salting effects.
Additional important concepts from the file
1) Specific vs non-specific elution
This is one of the core theoretical ideas in the lecture and easy to miss.
Specific elution
This means you release the protein by adding a molecule that directly competes for the same binding site.
Examples:
- imidazole competes with His-tag in IMAC
- NAD+ competes for enzymes binding immobilized NAD+
- salt ions compete in ion exchange
This is usually the preferred method, because it is more selective.
Only proteins using that exact interaction are eluted.
Non-specific elution
This means you disturb the binding environment more generally.
Examples:
- changing pH
- adding urea
- adding guanidinium chloride
- changing ionic strength
This weakens binding for many proteins at once.
So multiple proteins may come off.
This is less selective.
2) Competitive elution with cofactors (very important theory)
The lecture gives the NAD+ example, which is conceptually very important.
principle
If an enzyme naturally binds a cofactor like:
- Nicotinamide adenine dinucleotide (NAD+)
- ATP
- FAD
you can immobilize that cofactor on the column.
Then enzymes that recognize it will bind.
why can this cause competitive desorption?
Exactly because free NAD+ in solution competes with immobilized NAD+.
Like this:
" enzyme + column-NAD+ ⇌ bound "
then add free NAD +
"
enzyme + free NAD + ⇌ released "
The enzyme often prefers whichever interaction is more favorable under the conditions.
So yes — your interpretation was correct:
the coenzyme is attached to the support and free coenzyme is used to elute
3) Why low KD is so useful in purification
This is a major conceptual point.
Low KD means:
K_D < 10^{-6},M
very strong binding
This matters because purification columns often process large volumes of dilute sample.
Example:
- bacterial lysate
- serum
- cell culture supernatant
If affinity is weak, the protein leaks during washing.
So chromatography is really a balance between:
- strong enough to capture
- weak enough to release
This balance is one of the central themes of the file.
4) Why recombinant tags are so important
The lecture makes an important distinction here.
Modern labs often purify recombinant proteins
This means the protein is genetically engineered.
Example:
" protein + His-tag "
This dramatically simplifies purification.
Without a tag, purification often needs several steps:
- ion exchange * HIC * SEC
So tags reduce purification complexity.
This is a very important practical takeaway.
5) Charge distribution matters more than total charge
This is one of the most important conceptual ideas in ion exchange.
A protein may have many charged residues overall, but what matters is:
- are they on the ** surface **?
- are they ** accessible **?
- are they already involved in ** salt bridges **?
This is why pI predictions are helpful but imperfect.
Two proteins with similar pI can behave very differently on IEX.
Because:
- surface accessibility differs
- local charge clusters differ
- shape differs
- local charge clusters differ
This is a major reason chromatography remains partly empirical.
6) Charge density vs total charge
This was only briefly touched in your earlier question but is worth emphasizing.
Charge density means:
** how concentrated charges are in accessible regions **
A protein with 10 negative charges spread out may bind weaker than one with 6 tightly clustered negative charges.
Because clustered charges create stronger collective attraction.
This is why proteins with similar pI may elute at different salt concentrations.
7) Column chemistry stability
Very important overlooked concept.
The lecture distinguishes ** strong vs weak ion exchangers **
This does ** not ** mean stronger protein binding.
This is easy to misunderstand.
It refers to ** stability of the functional group across pH **
For example:
- strong exchanger = remains charged across broad pH range
- weak exchanger = only charged in narrower pH range
This affects usable buffer conditions.
Very exam - relevant concept.
8) Why HIC is often used after IEX
This is an important workflow idea implied by the file.
Because proteins often come off IEX in ** high salt **
that sample is already perfect for HIC loading.
HIC requires high salt to bind.
So these two methods often fit nicely in purification pipelines.
Typical workflow:
" IMAC → IEX → HIC → SEC "
or
" IEX → HIC → SEC "
This is a very practical purification design principle.
9) HIC preserves native structure
This is a big theoretical advantage.
Unlike reversed - phase chromatography, HIC is usually mild.
Proteins often remain folded.
That makes it suitable for:
- enzymes * binding proteins * structural studies
This is important if activity must be preserved.
10) Size exclusion chromatography(SEC) introduction
The file begins introducing this at the end.
This is different from all previous methods.
Previous methods rely on:
- affinity
- charge
- hydrophobicity
SEC relies on ** size / hydrodynamic radius **
Large proteins elute first.
Small proteins elute later.
This is because small proteins enter pores in the beads.
Large proteins cannot.
So they take a shorter path.
This principle is extremely important in protein chemistry.
Big - picture summary of the lecture
This lecture is really about one central idea:
proteins can be separated by exploiting different physicochemical properties
namely:
- ** specific binding **
- ** charge **
- ** surface hydrophobicity **
- ** size **
That framework is more important than memorizing each column.
Once you understand this logic, most purification schemes become intuitive.